您好,欢迎访问大连双航化学有限公司
语言选择:  ∷
∷ 
∷ ∷ 摘要:氟核磁共振光谱的使用为控制工艺过程和分析反应产物提供了独特的机会,其生产技术是在联邦国立统一企业“俄罗斯应用化学研究中心”的彼尔姆分公司开发的。本文介绍了生产四氟环氧乙烷,低聚物和基于它们的产品的已知方法。给出了使用核磁共振波谱法对氢和氟原子核控制工艺过程的详细信息。
关键字:氟19 NMR光谱;多氟化合物,TFE,氧化物,四氟环氧乙烷。
接收各种抗紫外线和辐射暴露的热稳定且耐腐蚀的材料需要开发用于合成和分析化合物的方法。其中,具有所需性能的聚合物和复合材料的全氟化含氧前体是第一位的。基于四氟环氧乙烷-单体-4氧化物(OTFE,OM-4,全氟环氧乙烷)的低聚物的全氟烷基乙烯基醚是非常重要的,其例如用于获得防冻橡胶。在本文中,我们分析了一系列基本上包含四氟环氧乙烷(-CF 2 CF 2O-),其生产已在联邦国家单一企业(彼尔姆分公司)“俄罗斯应用化学研究中心”的基础上掌握。该出版物给出了特征性的NMR化学位移(CS)(相对CFCl 3)用于分析。来自“分析”基团的信号使得可以确认混合物中是否存在化合物,并通过剩余部分的积分强度“组装分子”。前面我们已经描述了实验方法和组成计算[1-3]。反应混合物中的溶剂或各种化合物有助于氟NMR谱线的位置变化在3-5 ppm之内,但是,足够的位移范围(〜400 ppm)使得可以正确选择分析线并确定比率确定的组件。目标化合物中的杂质取决于生产方法。让我们考虑OTFE生产及其低聚的主要已知反应。
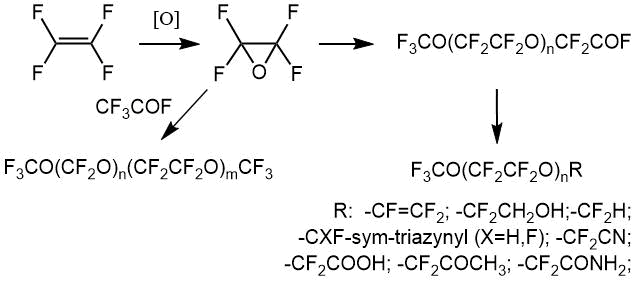
工业上以气相和液相形式生产四氟环氧乙烷(OTFE)。气相方法的特点是易于实现和仪器化。
气相方法之一是在大气压力和0至50°C的温度范围内,通过分子氧在氧化硅上氧化四氟乙烯(TFE)的过程(在190-300°C的温度下预混合气体) [4]);在气相中(波长范围为1800至3000Å,温度范围为-30至+ 150°C,压力为2 atm [5-6]的紫外线照射)。 ),同时形成大量的气态产物(碳氟化合物,全氟环丙烷,碳酸酐),液体产物(聚氧全氟亚甲基)和固体副产物。
以前,描述了在自由基源:氟,氯,溴,碘,氧化物和二氧化氮,溴化氢和三氟甲基次萤石的存在下用氧气对四氟乙烯进行光化学氧化的方法,该方法产生催化量的气态自由基。将氧气与惰性气体稀释剂预混合。自由基试剂的使用量很小,例如,气体混合物中的溴浓度在4·10 -5 至6·10 -5体积%的范围内。为了减少该过程的爆炸性,将氧气与惰性稀释剂(例如氮气)以1:1的比例预混合。氧与氟代烯烃的比例优选等于1:1 [6]。
液相氧化工艺在技术上更安全。
已知在0至40℃的温度范围内,在全氟化或氯氟化溶剂中并在臭氧的存在下,通过分子氧氧化四氟乙烯(TFE)的方法。副产物有羰基氟(COF 2)和全氟环丙烷。同样已知的方法是在存在以催化量产生的自由基源的情况下,通过液相中的氧气氧化TFE。氯,溴,碘,一氧化二氮/溴化氢和溴化氢被用作自由基的来源。以及作为溶剂的多氟烃和醚[6]。
以前,描述了在自由基源存在下通过氧气进行液相TFE氧化的方法,该方法使用氟代三氟甲烷(CF 3 OF)或元素氟,且该过程在15至60°C的温度范围内进行氟利昂318(全氟环丁烷)中的C。将引发剂以1-3%vol的量加入到氮-氧混合物(N 2:O 2= 1∶1)中。TFE:O 2的比例保持在1。在惰性氟代氯烃溶剂中,分子氧在分子氟的作用下进行四氟乙烯的液相氧化,同时搅拌,UV辐射并用作双(氟氧基)二氟甲烷的氧化引发剂[7]。 。
在实验室条件下,通过在分子氟的存在下用分子氧氧化TFE制备OTFE [8],在-50至125°C的温度范围内用气相氧气和UV辐照[9],并在空气中用臭氧氧化描述了在0°C [10-11]下的1,1,2-三氯-1,2,2-三氟乙烷。显然,反应通过形成摩尔-臭氧化物进行,随后形成羰基二氟化物和二氟环氧乙烷。四氟环氧乙烷也可以通过在氢氟酸中对环氧乙烷进行电化学氟化而制得[6]。
据报道,OTFE作为六氟-1,3-丁二烯与氮氧化物(IV)NO 2的自由基反应产物之一,在27至160℃的温度范围内获得。在二氧化氮对烯烃碳的攻击产物中 ,鉴定了CF 3 C(O)F,C(O)F 2,NO,FNO,SiF 4(来自玻璃)。TFE可以在相似的条件下反应[12]。
因此,先前已经很好地研究了各种制备四氟环氧乙烷的方法。在联邦国家统一企业“应用化学俄罗斯研究中心”的现场,根据方法[6,7]无需中间分离即可获得四氟环氧乙烷,并进一步转化为目标产物。NMR光谱通过色谱分析和IR光谱得到补充。来自反应器的气体混合物的组成通过GLC方法经由LCM-72设备进行监测,所述检测器-测温仪和色谱柱长5m,直径4mm,填充有硅烷化的硅色素。载气(氦气)的进料速度为1.5-2 l / h,柱温为20°C。此外,产品成分通过红外光谱确定,– 1,OTFE [6]的特征。
为了得到全氟聚醚羧酸氟化物,将四氟乙烯气体,氧气和气态或液态引发剂(一种或多种引发剂)供入含有液相溶剂的反应器中。或者,可以在工艺开始之前将引发剂(一种或多种引发剂)与液相完全引入反应器中。例如,当引发剂在室温下为液态时,可以使用此选项。作为反应的结果而获得的全氟聚醚过氧化含有式perfluoroxyalkylene组分(СF 2 СF 2 O)和(СF 2 O)。组分的摩尔浓度(СF 2 СF 2O)通常为5至95%(最好为20至90%)。根据本发明的方法,通常生产过氧化的全氟聚醚,其中末端COF基团与非官能末端基团的比率非常低。该比例通常小于5%(优选小于2%)。所得产物的数均分子量通常为几百至数十万道尔顿,即500至100000 Da [13]。应当指出,有可能在一处设施上以工业方式获得整个产品线,主要是酰基氟(以下简称“低聚物”)和“封闭”聚醚[14]。
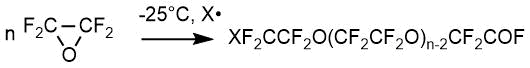
因此,相同或不同的端基表示以下基团:X F 2-,X F 2 F 2 -,-CFO和-F 2 F,其中X是引发剂和/或溶剂分子的片段。X通常为F,Cl或全氟烷基或全氟烷氧基。当引发剂包含两个OF键时,该片段可以连接到两个增长的聚合物分子上,并被掺入过氧化的全氟聚醚产物的分子链中。因此,取决于引发剂(一种或多种引发剂)和溶剂的性质以及工艺条件,端基的性质因产物而异。
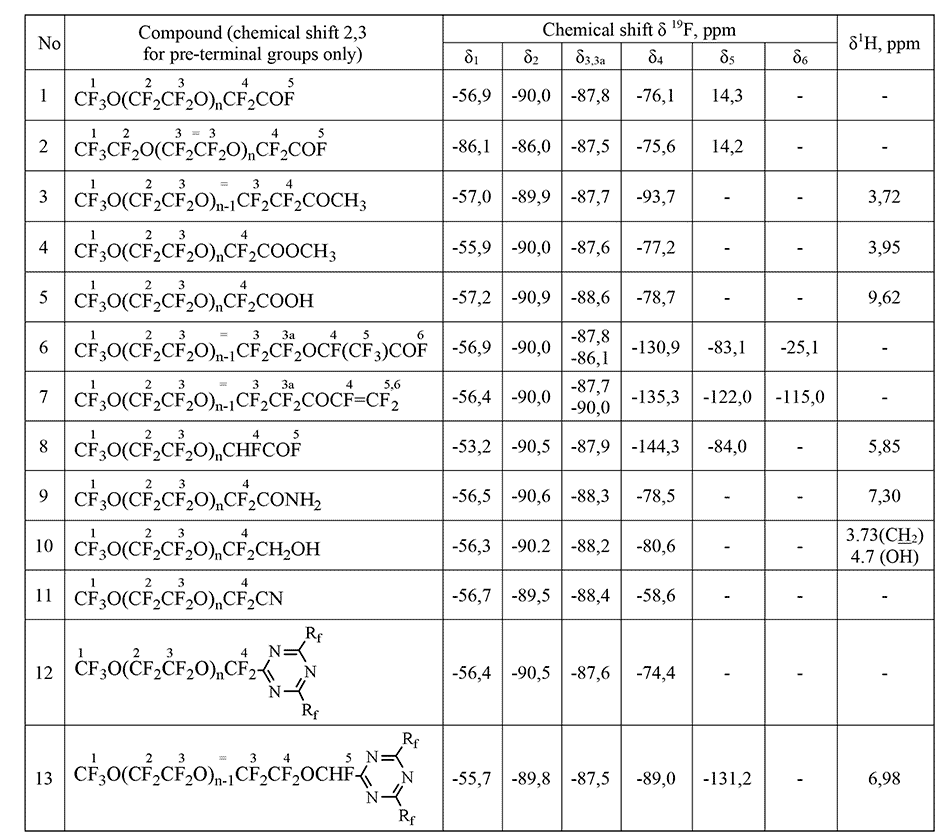
通过NMR方法进行定量分析的主要条件是存在至少一个吸收带,该吸收带不与其他吸收带重叠,并允许在制备的任何阶段从中间体或副产物的混合物中明确分离出该化合物。分析时间为25分钟,最大随机相对误差为1.2%。表2显示了低聚物的光谱特征,这些低聚物是在二甘醇二甲酸酯类型的质子惰性溶剂中OTFE与酸性氟化物反应的主要产物(因为杂质可能是OTFE与溶剂相互作用的产物-即相应的醇和简单醚)[15] 。这些低聚物,作为含反应性氟氢化物的物质,可以转化为各种衍生物:酸,酯,酰胺,醇,腈等。CF所有化合物共有的3 O(CF 2 CF 2 O)片段在与低聚物2相同的区域中具有光谱信号,但是,每种化合物都有其自己的特征谱带,因此可以以任何组分比率分析其混合物。当监测用于获得特定化合物的反应过程的完整性时,就是出现了这一任务,即必须能够分析通常包含几种组分的反应混合物。因此,例如,当监测酯12的生成时,各个阶段的反应混合物可能包含初始低聚物3,其杂质 5-7、9和最后的酯12。化合物3、6和 7具有接近的化学位移,但实践表明,化学位移δ(CF 2 COF)76 ppm <δ(CF 2 COOCH3)77 ppm <δ(CF 2 COOH)78 ppm的规律非常严格地观察到。
表1.四氟环氧乙烷低聚物及其衍生物的光谱特性。
制备全氟聚氧代烷基乙烯基醚的一般方案如下[16]:
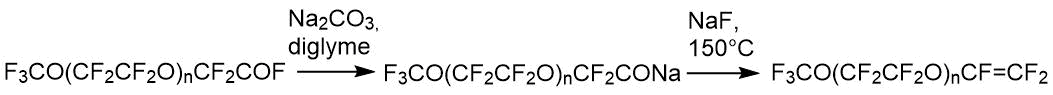
给定的反应链通过NMR在从初始到最终产物的所有阶段进行分析。对于所有化合物而言,CF 3 O(CF 2 CF 2 O)n碎片均常见,其特征在于19 F光谱中的吸收线位于光谱区域中的三重态-55÷-57 ppm。(CF 3 O-),前端四重奏-89÷-90 ppm(CF 3 OCF 2-)和内部片段的多重峰87 ppm(-CF 2 CF 2 O-)n。除了上面列出的吸收线外,每种化合物在19中都有自己的特征线F谱。因此,低聚物1在-76 ppm处具有对应于– CF 2 COF的光谱信号,化合物2的特征在于在–129 ppm(-CF(CF 3)-)处的比吸收带,而最终的酯3具有具有双键的三个氟是一组在-115,-122,-135 ppm的吸收线。这样的特征性非重叠信号的存在使定性成为可能(并且在比较信号的积分强度时),因此定量明确地确定了给定反应的主要产物和中间产物的组成。在我们的生产条件下,纯碱或二甘醇二甲醚中含氢杂质和水的存在导致形成副产物,如通式为F的氢化物3 CO(CF 2 CF 2)n CFHCF 3。使用PMR光谱法监测它们的存在,其中氢化物产生四重峰的四重峰(J H-F = 51.8 Hz,J H-CF3 = 2.7 Hz),吸收带为6.05 ppm。在19 F光谱中,对应于-CHF-基团的特征吸收带在-145ppm的光谱区域中,因此可以定性和定量地监测这种氢化物的形成。
氢化物可用作潜水泵的分离液。除通式4的氢化物外,还合成并分析了CF 3 O(CF 2 CF 2 O)n CF 2 H氢化物。这种氢化物的19 F光谱具有特征线–在-84.3 ppm的光谱区域中三重态的两倍。(J CF2-H = 67.0 Hz,J CF2-F = 6.0 Hz),在PMR光谱中-在6.45 ppm的光谱区域中为三重态。(J H- CF2≈67.0Hz)。
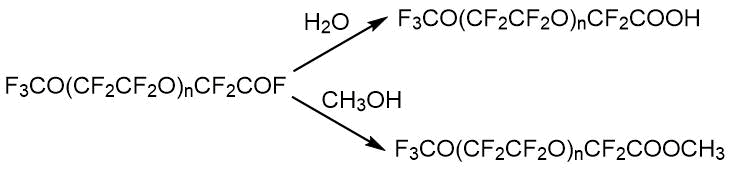
通过19 F NMR分析基于四氟环氧乙烷低聚物的n = 1-4的酸CF 3 O(CF 2 CF 2 O)n CF 2 COOH并不特别困难。对于任何n,酸在78.4 ppm的光谱范围内都有自己的特征线。在酸性样品中发现了它的甲酯以及游离的和结合的二甘醇二甲酸酯的残留物,这些以前没有被描述过[17]。由通式为CF 3 O(CF 2 CF 2 O)n-1 CH 3的醚形成醚7存在于起始低聚物中。通过特殊的实验证实了它的形成,包括对所得酸进行的各种处理以及处理后的起始低聚物和酸(1 H,CF 2 OCH 3 3.76 ppm)的NMR分析。平均而言,起始低聚物包含8摩尔。乙醚8。在酸性介质中制备全氟酸5时,-CF 2 CF 2 OCH 3转化为-CF 2 COOCH 3 光谱线消失,光谱线为-94 ppm,取代甲基羧基的信号出现为3.70 ppm。苛刻条件下的氟化作用不起作用,OCH 3含量保持在8%的水平。通过用硫酸处理可以完全除去二甘醇二甲醚,剩下约5%的酯。
另一种商业产品是全氟聚醚羧酸的甲酯。酸性甲酯样品的光谱清楚地显示了醚的存在,该醚是在初始低聚物的生产过程中获得的(由于与二甘醇二甲醚的相互作用)。对甲酯蒸馏各馏分的分析表明,混合物在111°С处含有最大量的醚和诸如二甘醇二甲醚的杂质,在130°С处,醚的光谱线消失。
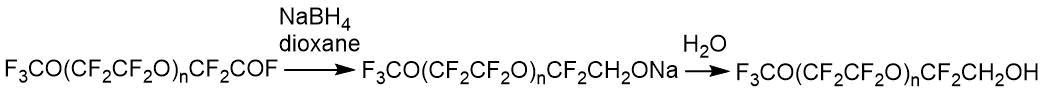
目标醇可用于生产丙烯酸酯。通过19 F NMR分析n = 1-5的醇。
表2.所分析醇主要基团的NMR化学位移。
|
组 |
化学位移,ppm |
|
-CH 2 - |
1小时:3.73吨 |
|
-哦 |
1 H:4.7(可能与杂质相互作用) |
|
-(CF 2 CF 2 O)n- |
19楼:-88,2 |
|
-CF 2 CH 2 - |
19楼:-80,6 |
|
CF 3 O- |
19楼:-56,3 |
|
CF 3 OCF 2 O- |
19楼:-90,2 |
在第一阶段中,在水的存在下,可以形成相应的酸,该酸固定在-77.6 ppm的光谱范围内。(-CF 2 COOH)。-OH基团信号的异常位置也可能表明正在研究的混合物中存在酸性介质。在随后水解的产物中,注意到形成特征化学位移为-75.6ppm的三氟乙酸。因此,在一些实验中,其量达到12%摩尔。

在一系列有机氟化合物中,通过在化学引发剂的存在下通过分子氧将四氟乙烯氧化而得到的,具有聚醚I和II的结构的含氧衍生物 已经普及。
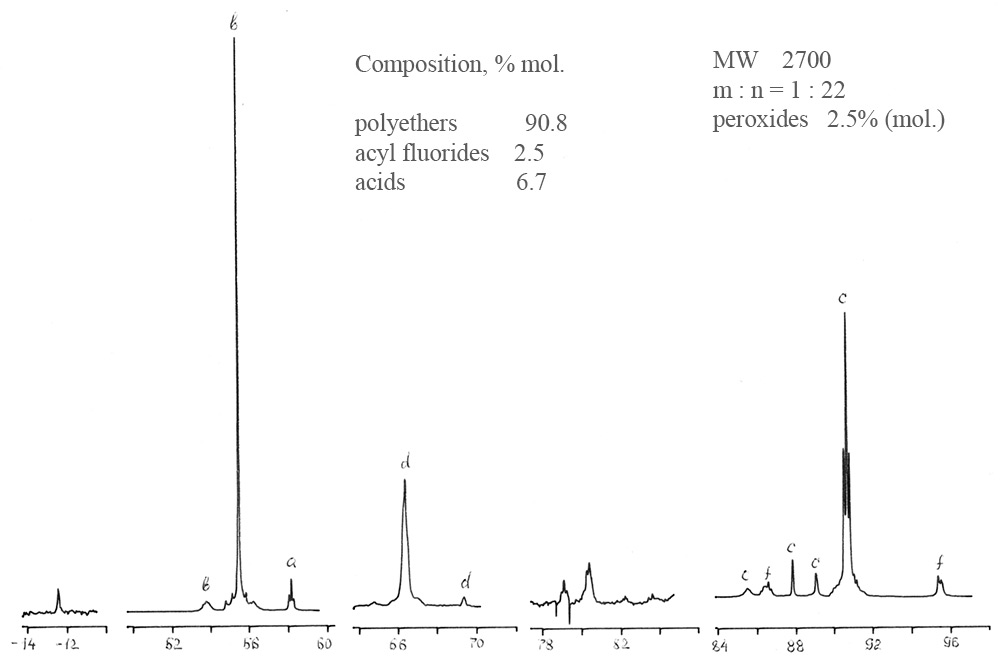
图1.粗聚醚I的19 F NMR光谱。
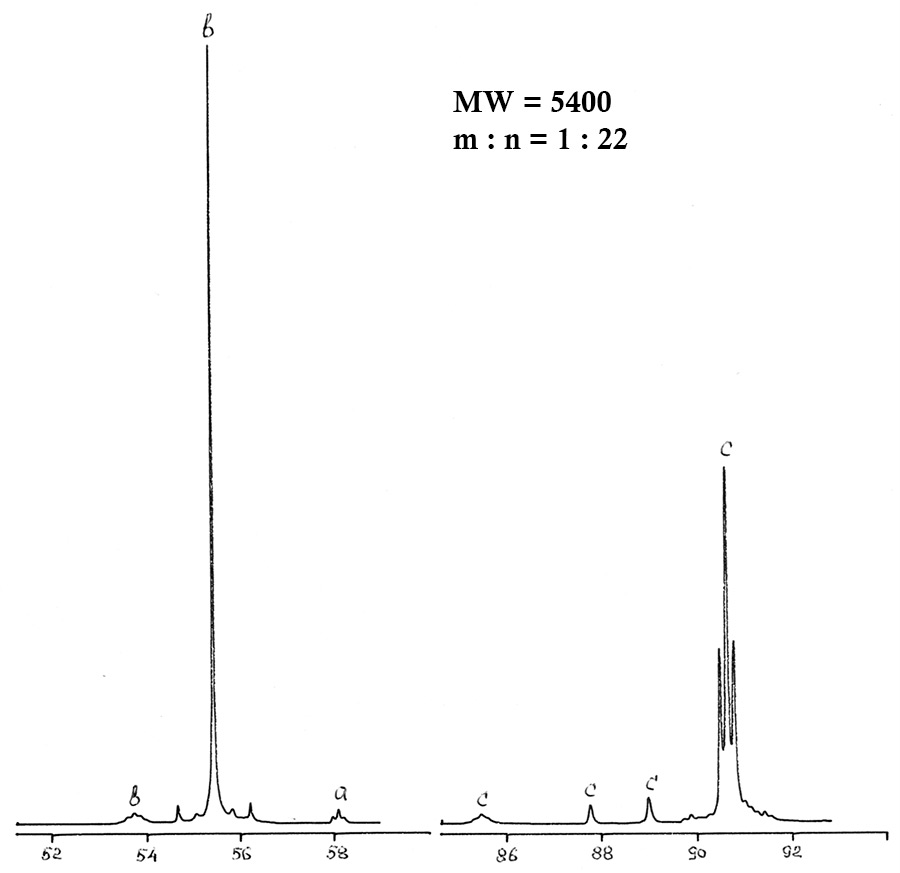
图2.处理后的聚醚II的19 F NMR光谱。
实际实践表明,传统的分析方法-色谱,红外光谱,化学分析-无法提供有关聚醚的详尽信息,也无法确定其分散的组成和合成监测。19 F NMR分析可以确定片段组成,平均分子量和含有过氧化物(活性)氧的组分的百分比。在分子量的测量范围内以及在102至104 Da(amu)的密度范围内,总相对误差为7%。表3给出了聚醚II 型CF3O(CF2O)m(O)x(CF2CF2O)n(O)y CF3的光谱线分配 。
表3. 分析的聚醚主要组的19 F化学位移。
|
组 |
19 F 化学位移,ppm |
频谱名称 |
|
-CF 2 O- |
-52,7÷-54,4 |
b |
|
-CF 2 CF 2 O-O- |
-85,2÷-94,4 |
F |
|
-CF 2 O-O- |
-64,2÷-66,2 |
d |
|
CF 3 O- |
-57,2 |
一种 |
|
CF 3 CF 2 O- |
-86,0÷-89,6 |
C |
聚甲醛(POM)生产技术包括在freon-318介质中生产分子量为2400-3400 Da的原油,并进行溶剂汽提,热稳定和(在某些情况下)化学处理以减少过氧化物和酸的量氟化物,以增加目标聚醚的稳定性和惰性。在聚醚合成我形成了下一个功能性化合物-酰基氟和酸,通过氟处理将其转化为“封闭”结构。通过在-79÷-80至+12.4 ppm范围内的谱线消失来控制该处理的完整性。氟处理还可去除其他含氢杂质,定性记录在PMR光谱中。
核磁共振波谱学用于研究分子量对单体消耗量的依赖性。发现上述间隔中的依赖性是线性的。
因此,基于19 F NMR方法的分析以及PMR光谱的补充(考虑到其可操作性和信息性),可以控制获得各种聚醚及其衍生物的过程,控制目标产物的收率,并且-快速优化技术。
Copyright © 2014-2022 大连双航化学有限公司 版权所有 地址:中国.大连市经济技术开发区红星海B区207号1-16 备案号:备202101188号 网站地图
![1.1.2.2-四氟-2-[(1.2.2-三氟乙烯基)氧基]乙烷](/uploads/210104/1-2101041AZ1951.png)
![5.8-二氢吲哚并[2.3-c]咔唑](/uploads/210104/1-2101041AHbY.png)


